













AlphaFold和RoseTTAFold雙劍合璧,北大校友領銜登上Science
本文經AI新媒體量子位(公眾號ID:QbitAI)授權轉載,轉載請聯系出處。
AlphaFold和RoseTTAFold等AI的橫空出世,可以說打開了蛋白質預測新世界的大門。
而現在,依靠這倆AI模型的“組合拳”,科學家們又有了新的突破:
首次確定了超過100種“疑似”全新蛋白質復合體,并為700多種此前結構未知蛋白質復合體提供了3D結構預測。
也就是說,現在,AI現在不僅能預測蛋白質單體,還能成功預測蛋白質之間的相互作用了。
研究人員興奮地表示:
我們的結果是結構生物學新時代的一個重大進展。
在這個時代,計算將在結構生物學中起到根本性的作用。

這項研究來自德州大學西南醫學中心和華盛頓大學領銜的國際團隊。
論文已經發表在《Science》上。
“結構生物學新時代的重大進展”
我們知道,蛋白質通常以復合物的形式成對或成組地發揮功能,以完成生物體生存所需的種種任務。
但現今為止,許多真核生物蛋白質復合物的結構仍然成謎,其中蛋白質之間的相互作用也尚未被識別。
而這篇Science論文完成的主要工作,就是將基于深度學習的蛋白質預測方法,引入到了蛋白質復合體的研究當中。
具體而言,研究人員利用全蛋白質組氨基酸協同進化分析和“RoseTTAFold + AlphaFold”的組合,系統地識別和建立了真核生物核心蛋白質復合物的精確模型。
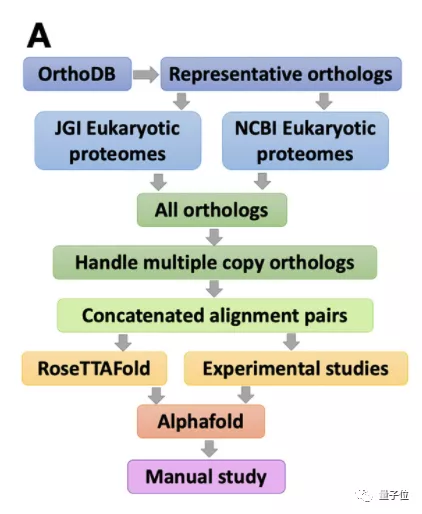
RoseTTAFold和AlphaFold的作用,都是根據氨基酸序列預測蛋白質的3D結構。
有所不同的是,RoseTTAFold速度更快:采用了3軌注意力機制,分別關注蛋白質的一級結構、二級結構和三級結構;再通過在三者之間加上多處連接,使整個神經網絡能夠同時學習3個維度層次的信息。
此次的論文還提及,RoseTTAFold團隊設計了一個雙軌道模型,雖然準確率有所損失,但在計算時間上要比AlphaFold快100倍。
而通過實驗,研究人員認為,采用雙軌注意力機制的AlphaFold在預測蛋白質復合體方面會有更高的準確率。
因此,在這項研究中,研究團隊結合了兩者的優勢:
首先在酵母菌的基因組中尋找以相互關聯的方式獲得突變的基因。
然后,利用上述兩種AI技術確定這些蛋白質是否可以組合成3D結構。
研究人員最終從酵母菌里篩選出了830萬對蛋白質,并從中識別出1505種可能的蛋白質復合體。
其中699個蛋白質復合體的3D結構已經在此前的實驗中被解析出來。
剩下的806個中,有實驗數據支持的預測結果有700個,另外106種屬于從未被描述過的全新蛋白質復合體。
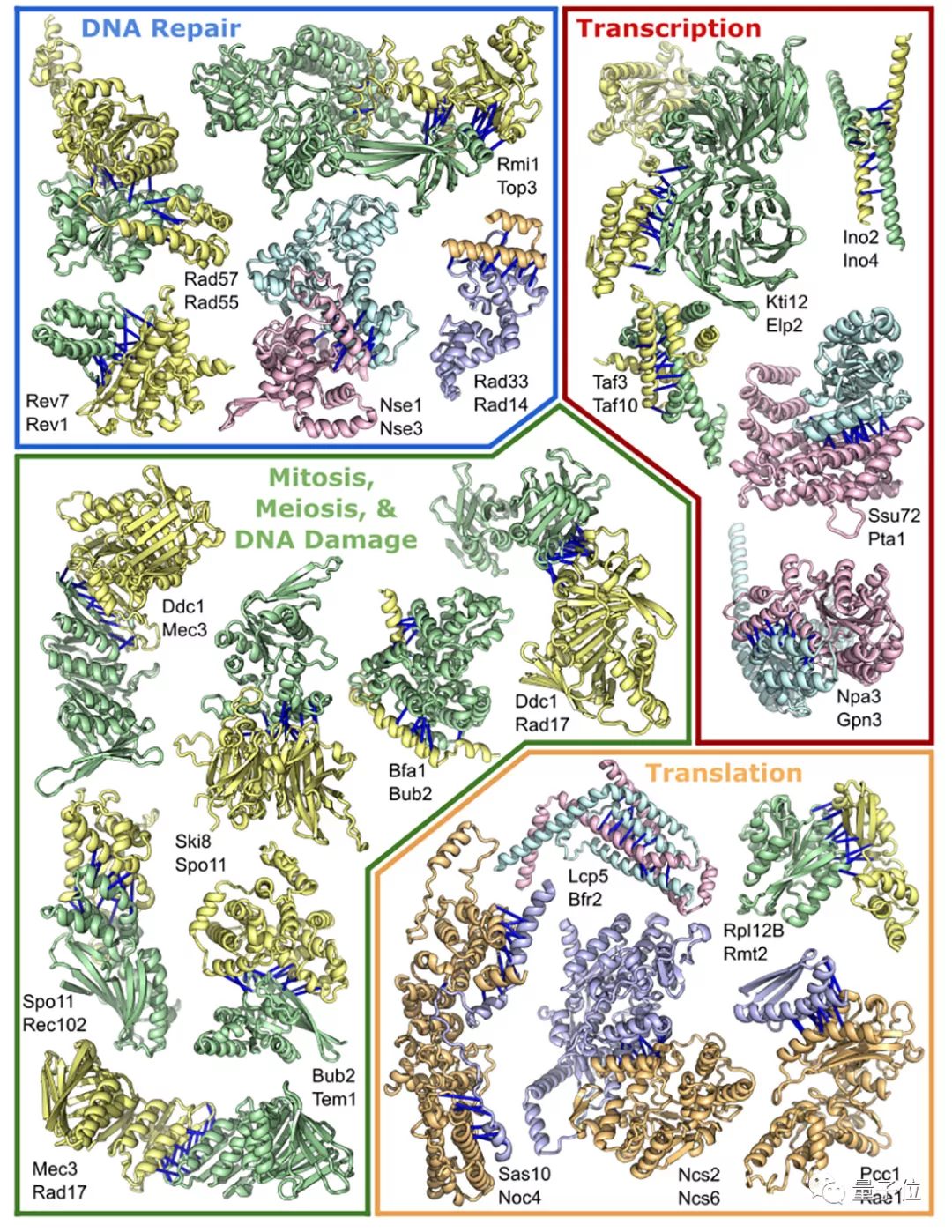
這項工作為類似的人體蛋白質相互作用研究奠定了基礎,最終可能有助于開發治療人類疾病的新療法。
華人領銜的研究團隊
這項研究由德州大學西南醫學中心助理教授叢倩團隊和華盛頓大學蛋白設計研究所教授David Baker團隊聯合發表。
論文共同通訊作者叢倩本科畢業于北京大學。2017年-2020年在華盛頓大學David Baker教授門下任博士后。
她也是RoseTTAFold的作者之一。
論文另一位通訊作者David Baker是美國生物化學家和計算生物學家,現為華盛頓大學生物化學教授、華盛頓大學蛋白質研究所貝克實驗室首席研究員。
正是他帶領的團隊一手打造了和AlphaFold2一起沸騰學術圈的RoseTTAFold。























